
Das Herpes-Simplex-Virus 1 (HSV-1) ist eines der Viren, die die meisten Menschen lebenslang im Körper tragen. Nach der Infektion im Kleinkindalter „schläft” das Virus in spezialisierten Zellen in unserem Körper, bei HSV-1 sind das Nervenzellen im Gesicht. Diese Zellen leiten bspw. Empfindungen – Berührungen, Hitz – von unseren Lippen ins Gehirn. Manchmal kann das HSV-1-Virus aber auch „aufwachen“, und sich wieder vermehren. Es „reaktiviert“ in den Nervenzellen und wandert in Richtung Lippen. Dort ist die schnelle Vermehrung des Virus dann als Fieberblase sichtbar und spürbar. Das ist schmerzhaft und lästig, lässt sich aber mit entsprechenden Salben behandeln. Manchmal wandert das Virus aber in die falsche Richtung, ins Gehirn*. Dort kann es eine lebensbedrohliche Hirnentzündung verursachen – davon gibt es in Deutschland einige Hundert Fälle pro Jahr. Und auch wenn eine schnelle Behandlung das Leben der Patientinnen und Patienten rettet, treten meistens bleibende Spätfolgen auf wie epileptische Anfälle. Grund dafür sind wahrscheinlich beschädigte Nervenzellen. Die Frage ist nun, warum eine HSV-1-Gehirnentzündung zu diesen Spätfolgen führt, und wie sie verhindert werden könnte.
Eine Virusinfektion erforscht man am besten mit Proben aus Menschen: also bspw. etwas Gewebe oder Blut zu entnehmen, und mit verschiedenen Experimenten feststellen, welche molekularen und zellulären Prozesse stattfinden. Praktisch ist das aber oft kaum möglich. Gerade aus dem Gehirn können nur in seltensten Fälle Proben entnommen werden. Und vor allem nicht für wissenschaftliche Forschung – wenn, dann nur für ein Diagnoseverfahren, das der betroffenen Person direkt zu Gute kommt. Es braucht daher sogenannte „Krankheitsmodelle“: ein experimentelles System, mit dem wir die Hirnentzündung nach HSV-1-Infektion nachbilden können. Und hier kommen die Minihirne ins Spiel, die wir in unserer Publikation „Modelling viral encephalitis caused by herpes simplex virus 1 infection in cerebral organoids“ untersucht haben.
Von Zellen zu Organoiden
Schon seit Jahrzehnten wird in der Forschung mit menschlichen Zellen gearbeitet. Oft ist das aber nur ein bestimmter Typ von Zellen, die in einer flachen Plastikschale wachsen – nicht zu vergleichen mit einem menschlichen Organ, in dem Dutzende verschiedene Zelltypen in einer dreidimensionalen Struktur organisiert sind. Um das abzubilden, sind in den letzten 10 Jahren die „Organoide“ entwickelt worden. Hirnorganoide etwa, „Minihirne“ sind kleine Kügelchen, ein bis drei Millimeter groß, in denen sich Millionen verschiedene Zelltypen ähnlich wie in unserem Gehirn anordnen.
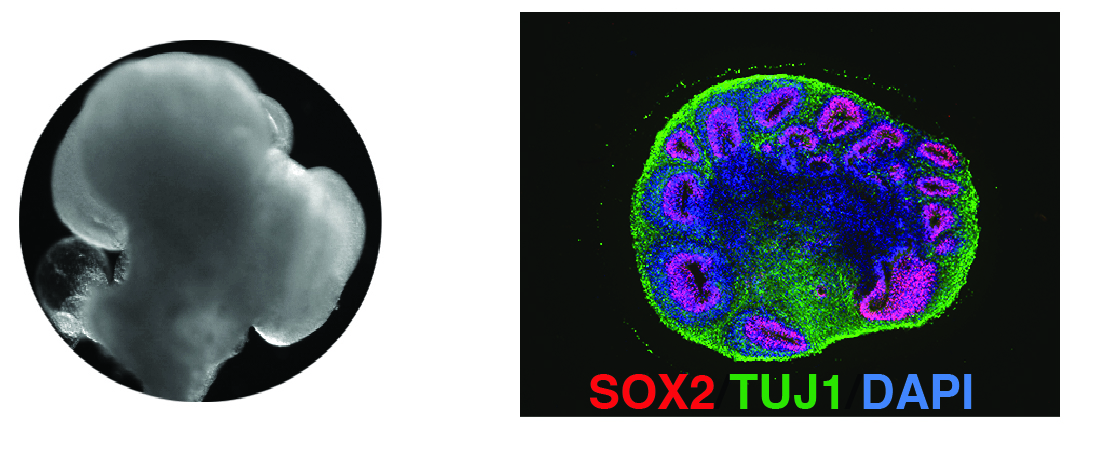
Auch wenn zu einem richtigen Gehirn noch sehr viel fehlt: Mit den Hirnorganoiden haben wir also nun ein deutlich besseres Krankheitsmodell für Herpes-Hirnentzündungen, und können im Labor untersuchen, wie die Infektion die Nervenzellen schädigt.
Gute und schlechte Seiten des Immunsystems
Im Labor haben wir dann die Hirnorganoide mit dem Virus infiziert. Gegen HSV-1 gibt es mit Aciclovir ein jahrzehntealtes, wirksames Medikament. Auch in den Hirnorganoiden hat Aciclovir das Virus gut an der Vermehrung gehindert. Trotzdem litten die Nervenzellen in den Organoiden, auch wenn sie gar nicht infiziert wurden. Einen Hinweis darauf gab die Einzelzellsequenzierung. Mit dieser Methode können wir für Tausende einzelner Zellen in den Hirnorganoiden feststellen, welche Gene sie in Folge der Infektion wie stark aktivieren. Damit sahen wir, dass nach dem Kontakt der Organoide mit dem Virus ein Teils des angeborenen Immunsystems angeschaltet wird. Dieser sogenannte TNF-Signalweg trägt dazu bei, eine Entzündung – also die notwendige Aktivierung des Immunsystems zur Bekämpfung des Virus – auszulösen. Aber, für die Nervenzellen kann das problematische Folgen haben. Wir prüften also, ob das Bremsen dieses Signalweges den Nervenzellen in den Organoiden helfen würde – und das war tatsächlich der Fall. Vor allem die kombinierte Behandlung, also Aciclovir gegen die Virusvermehrung und ein Medikament gegen den TNF-Signalweg, „rettete“ die Hirnorganoide **.
Und jetzt?
Ganz überraschend sind unsere Erkenntnisse nicht, und es gab tatsächlich auch schon Versuche in der Klinik, entzündungshemmende Medikamente bei Herpes-Hirnentzündung anzuwenden. In der Praxis ist das nicht so einfach, weil „entzündungshemmend“ immer bedeutet, dass das Immunsystem auch daran gehindert wird, das Virus zu bekämpfen. Das gilt vor allem für häufig angewendete, breit wirksame „Entzündungshemmer“ etwa aus der Familie der Corticoide. Solche detaillierten molekularen Untersuchungen wie unsere können nun viel exakter definieren, was genau im Immunsystem den Schaden verursacht. Mit diesen Erkenntnissen kann man dann versuchen, genau passenden Medikamente zu finden. Im Hinblick darauf zeigt unsere Publikation auf, wie wertvoll Hirnorganoide für diese Forschung sein können.
Modelling viral encephalitis caused by herpes simplex virus 1 infection in cerebral organoids
Agnieszka Rybak-Wolf, Emanuel Wyler, Tancredi Massimo Pentimalli, Ivano Legnini, Anna Oliveras Martinez, Petar Glažar, Anna Loewa, Seung Joon Kim, Benedikt B. Kaufer, Andrew Woehler, Markus Landthaler & Nikolaus Rajewskys. Nature Microbiology (2023)
https://www.nature.com/articles/s41564-023-01405-y
* Es ist nicht ganz klar, ob diese Hirnentzündungen durch Reaktivierungen des Virus verursacht werden, oder durch Neuinfektionen. Ersteres wird als wahrscheinlicher angenommen.
** Wem das bekannt vorkommt: bei COVID-19 haben wir einen ähnlichen Effekt festgestellt. Dort war das Bremsen der Entzündung alleine noch deutlich effizienter als hier beim Herpes-Simplex-Virus, siehe https://emanuelwyler.wordpress.com/2021/08/18/coronainfo-funfter-teil/#1222.
Vor drei Jahren begann unsere Forschung am Coronavirus SARS-CoV-2, das die Krankheit COVID-19 auslöst. Ein großer Teil unserer Arbeit dreht sich um die molekularen Prozesse nach einer Ansteckung mit dem Virus. Auch wenn erst einmal der reine Erkenntnisgewinn im Vordergrund steht: gerade bei so akuten Themen stellt sich immer die Frage, ob und wie man all dieses Wissen denn anwenden kann. Und eigentlich klingt das ja auch ganz einfach: wenn man genau weiß, was nach einer Ansteckung geschieht, kann man mit Medikamenten so eingreifen, dass das Virus möglichst wenig Schaden anrichten kann. Das ist auch der Weg, den wir vor drei Jahren eingeschlagen haben – im Wissen darum, dass die Realität viel komplizierter ist, es viel zu lernen gibt, und sehr viele Möglichkeiten zu scheitern.
Aus dem ersten Projekt im Frühling 2020 folgerten wir, dass ein Medikament, das das Enzym Hsp90 in menschlichen Zellen hemmt, vielversprechend sein könnte. Denn damit, so die ersten Experimente, könnten gleich beide der zentralen Ziele bei COVID-19 angegangen werden: erstens die Vermehrung des Virus zu bremsen, und zweitens die bisweilen überschießende Reaktion des Immunsystems auf das Virus. Denn so sehr eine solche „Entzündung“ stattfinden muss, um das Virus in den Griff zu kriegen: was bei milder Erkrankung als „Erkältung“ spürbar ist, kann sich zu einer lebensbedrohlichen Lungenentzündung auswachsen oder zu Blutgefäßschäden führen – und auch bei Langzeitfolgen („LongCovid“/“PostCovid“) eine Rolle spielen.
Die ersten Experimente damals wurden in menschlichen Zellen gemacht – aber in einem künstlichen System, in Nährmedien in Plastikschalen. So anzufangen ist der natürliche erste Schritt, aber vieles, was unter diesen Bedingungen Erfolge verspricht, funktioniert im Menschen dann nicht (dazu gehören auch zu Pandemiebeginn so gehypte Dinger wie Remdesivir, Ivermectin oder Chloroquin). Die nächste Etappe in solchen Tests waren daher Tierversuche – um die Frage zu beantworten: funktioniert im ganzen Organismus, was in der Plastikschale Wirkung zeigt?

Um diese Frage zu klären, arbeiteten wir mit dem Institut für Virologie der Freien Universität Berlin, sowie der Firma Aldeyra aus den USA zusammen – letztere produziert einen Hsp90-Hemmer, der auch schon in klinischen Studien bei Menschen getestet wurde, insbesondere für verschiedene Krebsarten. Die Studie erschien kürzlich als „preprint“, d.h. in noch nicht begutachteter Form.
Als erstes hatten wir nochmals die Wirkung des Hsp90-Hemmers in menschlichen Lungenzellen getestet, und vor allem geschaut wieviel es braucht um die Virusvermehrung bzw. die Entzündung abzubremsen.

Etwas überraschend war, dass es mehr vom Hsp90-Hemmer Ganetespib braucht, um das Virus, als um die Entzündung zu bremsen. Das war wichtig zu wissen, denn später in den Tierexperimenten sahen wir, dass im Hamsterkörper die zum Bremsen des Virus notwendige Menge gar nicht oder nur kurz erreicht wird – das Medikament wurde wahrscheinlich zu schnell wieder ausgeschieden.

Mit dem Hsp90-Hemmer Ganetespib wurden die Tiere etwas weniger krank, die Menge Virus blieb aber, nicht unerwartet, gleich. Eine genaue Inspektion der Lunge zeigte tatsächlich auch eine in Maßen reduzierte Lungen- und Blutgefäßentzündung.
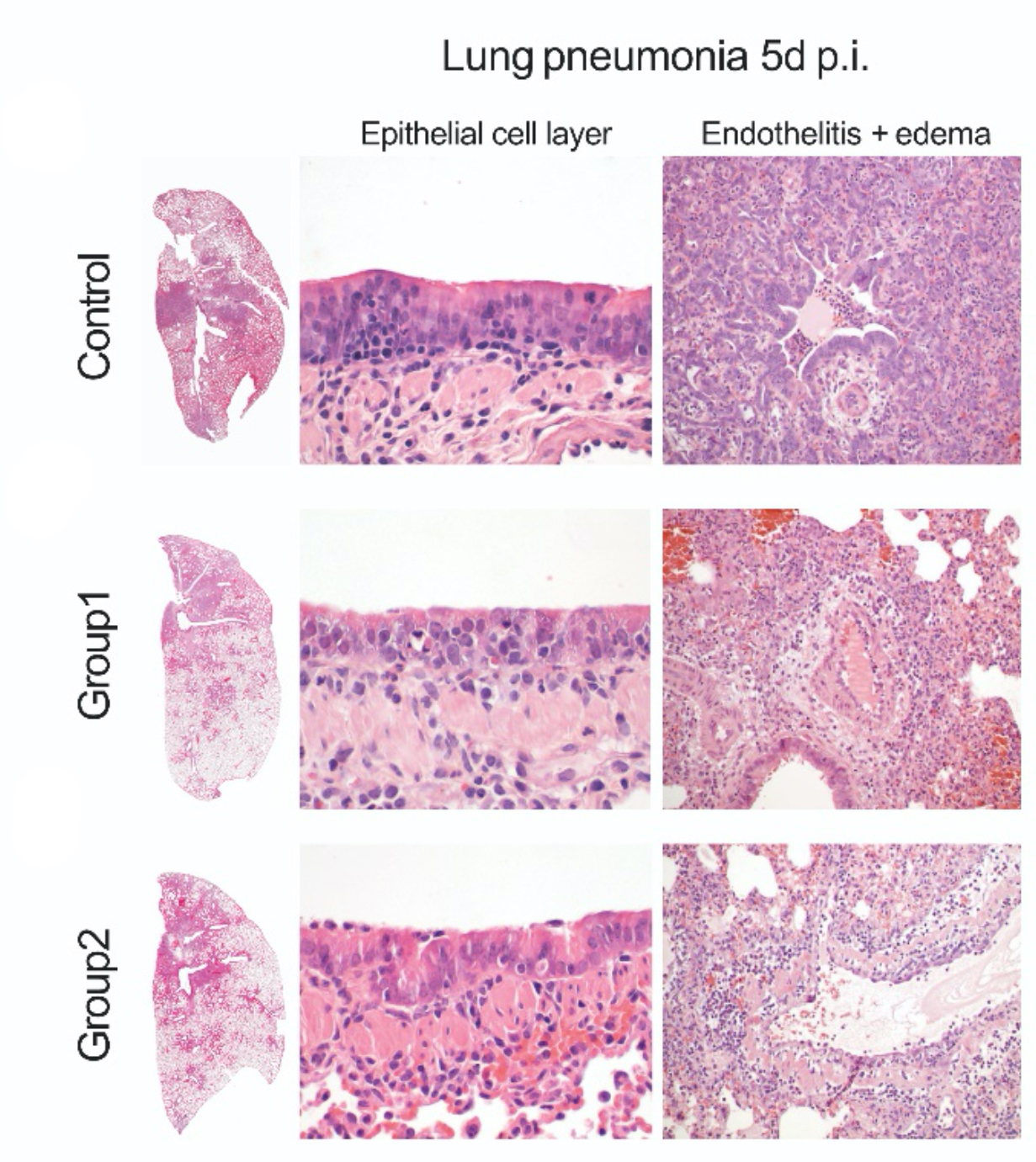
Wie das bei Menschen gebräuchliche COVID-19-Medikaments Dexamethason reduziert der Hsp90-Hemmer Ganetespib also nicht das Virus, wohl aber die Entzündung und damit die Krankheitsschwere. Der Effekt ist aber nicht sehr groß, und weiterhin ist unklar, ob das Medikament im Menschen etwas bewirken würde. Das Potenzial ist denn auch gar nicht so sehr als COVID-19-Medikament; weil es gegen Entzündungsmechanismen an sich wirkt und nicht gegen das Virus, könnte es bspw. auch bei Lungenentzündungen von Bakterien oder anderen Viren helfen. Daher werden wir die Möglichkeiten von Hsp90-Hemmern in Infektionskrankheiten im Rahmen des Covipa-Konsortiums der Helmholtz-Gemeinschaft weiter erforschen.
Die Google-Suche nach „Herpes“ ergibt 44 Millionen Resultate – kein Wunder, die Viren aus der Herpes-Familie sind weltweit verbreitet, und die meisten Menschen damit infiziert. Die wichtigsten Vertreter sind Herpes Simplex Virus 1 (verursacht Fieberbläschen an den Lippen, d.h auch „Lippenherpes“ genannt) und Herpes Simplex Virus 2 (Genitalherpes). Zu den Herpes-Viren gehört auch der Epstein-Barr-Virus, der die Mononukleose (Pfeiffer-Drüsenfieber) verursacht. Allen Herpesviren ist gemeinsam, dass sie eine sogenannte „latente“ Infektion verursachen, d.h. der Virus ist in einem Ruhezustand im Körper, und wird unter gewissen Bedingungen aktiviert zu einer „lytischen“ Infektion. Das ist bspw. dann der Fall, wenn bei Stress oder Erkältung wieder Fieberbläschen entstehen: der Virus hat seinen Ruhezustand verlassen, und die Zellen in der Lippe „lytisch“ infiziert. Bei gesunden Menschen ist das Immunsystem aber meistens in der Lage, lytische Infektionen zu verhindern.
Die Besonderheit von Viren ist, dass sie (im Gegensatz zu Bakterien) in die Zellen unseres Körpers eindringen. Obwohl Viren sehr einfach aufgebaut sind und nur wenige Gene haben, gelingt ihnen etwas so faszinierendes wie gefährliches: Viren können die hochkomplexen menschliche Zelle so beeinflussen, dass sie nur noch Viren produziert, und schlussendlich eingeht. Dazu manipuliert der Virus auch die Expression menschlicher Gene, das Forschungsgebiet unserer Arbeitsgruppe am Max-Delbrück-Centrum Berlin. Siehe dazu auch zwei ältere Blogbeiträge zu posttranskriptioneller Genregulation oder ringförmigen RNA-Molekülen.
In unserer neusten Publikation, „Widespread activation of antisense transcription of the host genome during herpes simplex virus 1 infection“, haben wir nun ein bisher unentdecktes Phänomen beschrieben, wie Herpes Simplex diese Genexpression manipuliert. Nach der Infektion von menschlichen Zellen in Zellkultur mit Herpes Simplex 1 haben wir gesehen, dass bei ungefähr 1000 von den 25000 menschlichen Genen sogenannte Antisense-Expression sichtbar wird.
Im Bild ist sehr schematisch und vereinfacht gezeigt, was das bedeutet. Unsere Gene sind auf den Chromosomen wie auf einer sehr langen Perlenkette aufgereiht. Im Bild ist mit einem orangen Pfeil ist der Start eines Gens bezeichnet. Dort wie die Information vom Chromosom als RNA-Molekül (=Transkript) abgelesen (=transkribiert), bis zum Ende des Gens. Dabei werden die Teile, die nicht sogenannte „Exons“ sind, herausgeschnitten. Von der RNA wird dann die Information in ein Protein übersetzt, das eine bestimmte Funktion in der Zelle ausübt. Das Antisense-Transkript, das nach der Herpes-Infektion entsteht, wird nun in entgegengesetzte Richtung abgelesen. Dabei kann es mit dem Gen überlappen (wie das untere, als „intern“ bezeichnet) oder auch nicht (sogenannt „divergent“). Nach der Herpes-Infektion werden also bei ungefähr 5% der menschlichen Gene neue Transkripte gebildet, und zwar in die dem Gen entgegengesetzte (englisch „antisense“) Richtung.
Das Antisense-Transkript, das nach der Herpes-Infektion entsteht, wird nun in entgegengesetzte Richtung abgelesen. Dabei kann es mit dem Gen überlappen (wie das untere, als „intern“ bezeichnet) oder auch nicht (sogenannt „divergent“). Nach der Herpes-Infektion werden also bei ungefähr 5% der menschlichen Gene neue Transkripte gebildet, und zwar in die dem Gen entgegengesetzte (englisch „antisense“) Richtung.
Die zwei grundsätzlichen Fragen sind:
1. Welches sind die Ursachen dieser Antisense-Transkripte?
2. Und welche Folgen hat die Transkription dieser Antisense-Transkripte?
Bei Frage eins sind wir zuerst der Möglichkeit nachgegangen, ob alle Virusinfektionen, oder allgemein Stressituationen für die Zelle, zu solchen Antisense-Transkripten führen. Während nach Infektion mit nahe verwandten Viren wie Herpes Simplex Virus 2 oder das Varizella-Zoster-Virus (Windpocken) zumindest ein Großteil der Antisense-Transkripte sichtbar ist, ist dies bei anderen Viren und einige unterschiedlichen Stresssituationen nicht der Fall. Für diese Analyse konnten wir auf publizierte Rohdaten anderer WissenschaftlerInnen zurückgreifen, die damit andere Dinge untersucht haben. Dies zeigt, nebenbei gesagt, wie wichtig es ist, dass in der Wissenschaft Daten zugänglich gemacht werden. Im nächsten Schritt versuchten wir einzugrenzen, welche Virusproteine die Antisense-Transkription verursachen. Dazu verwendeten wir mutierte Viren, die jeweils eines ihrer eigenen Proteine nicht herstellen können, sowie ein antivirales Medikament. Beides führt dazu, dass die Infektion langsamer verläuft, oder in einem frühen Stadium abbricht. Dabei bestätigte sich die Annahme, dass die ungefähr 1000 Antisense-Transkripte nicht alle dieselbe Ursache haben. Während für einige ein spezifisches virales Protein (ICP4, ein sogenannter Transkriptionsfaktor) verantwortlich ist, gelang bei andere Antisense-Transkripte nur eine ungefähre Eingrenzung der möglichen viralen Faktoren. Wahrscheinlich sind auch komplexere Mechanismen, bei denen verschiedene virale und menschliche Proteine zusammenwirken.
Die zweite Frage konnten wir ebenfalls nicht endgültig beantworten (aber dann wäre ja langweilig und es gäbe nix mehr zu tun). Ein angesichts früher Forschung naheliegendes Szenario ist, dass der Virus damit generell die Transkription vom menschlichen Genom „verwirren“ will. Denn einerseits sollen während der Infektion alle Ressourcen zu Transkription des viralen Genoms verwendet werden. Andererseits ist es für den Virus von Nachteil, wenn antivirale menschliche Gene transkribiert werden. Experimentell ist diese Hypothese schwierig zu belegen. Andererseits kann es sein, dass zumindest ein Teil der Antisense-Transkripte eine spezifische Funktion hat. Dazu haben wir das Antisense-Transkript zum BBC3-Gen näher untersucht. Das BBC3-Gen produziert ein Protein, dass sofort zu Apoptose führt (Selbstmord der Zelle). Es ist für die Virusinfektion von Vorteil, die Apoptose zu verhindern, und deswegen auch die Transkription des BBC3-Gens. Denn wenn die Zelle in Apoptose geht, kann sie keinen Virus mehr produzieren. Verschiedene unserer Experimente haben Hinweise gegeben, dass das BBC3-Antisense-Transkript die Produktion des BBC3-Proteins tatsächlich reduziert, und damit die Wahrscheinlichkeit für Apoptose reduziert.
Was ist nun die Relevanz dieser Arbeit? Für die Virusforschung ist interessant, welche tiefgehenden und eher unerwarteten Effekte ein Virus auf die Transkription haben kann. Dies ist ein weiterer Aspekt, wie Viren unsere Zellen in allen möglichen Bereichen manipulieren können. Für die Erforschung der Gen-Transkription sind die Liste der 1000 Antisense-Transkripte einerseits eine Vorlage, mit denen andere Datensätzen durchsucht werden können. Anderseits zeigen wir, wie Herpesviren als Methode verwendet werden können, um die Transkription in menschlichen Zellen zu verstehen, die in ihrer Komplexität wohl erst ansatzweise verstanden wird.
Eine faszinierende Frage in der Biologie ist: wie kann ein Haufen von biochemischen Molekülen, die sich nur so verhalten wie physikalisch-chemische Regeln vorschreiben, etwas so komplexes, kreatives, (manchmal) intelligentes, wie einen Menschen ergeben? Diese Frage wird hier nicht beantwortet, und auch nicht wie Krebs geheilt wird, aber ich erkläre unsere neuste Publikation. Darin beschreiben wir, wie menschliche Zellen Signale von außen unterscheiden können. Konkret geht es um den MAP-Signalweg, und genauer darum, wie die Zelle unterscheidet zwischen einem kurzen und einem langen Signalimpuls. Der MAP-Signalweg ist in der Krebsforschung wichtig, da er in vielen Tumoren übermäßig aktiv ist, und damit Zellwachstum fördert. In unserer Publikation, An immediate–late gene expression module decodes ERK signal duration, beschreiben wir, wie die Stabilität von RNA-Molekülen wichtig ist für die Signal-Dekodierung. Dies gehört ebenfalls zum Forschungsfeld der post-transkriptionellen Gen-Regulierung, die ich hier schon eingeführt habe. Die Publikation ist ein Zwischenschritt in einer längerfristig angelegten Zusammenarbeit zwischen Forschungsgruppen am Max-Delbrück-Centrum under Charité im Rahmen des Berliner Instituts für Gesundheitsforschung / Berlin Institute of Health.
Als Modell für die Experimente verwenden wir Zellen, in denen wir einen Faktor für den MAP-Signalweg anschalten können. Dieser Faktor, das RAF-Protein, aktiviert den Signalweg. Durch Zugabe eines Stoffes, U0126, kann der Signalweg wieder ausgeschaltet werden. Bekannt ist, dass ein kurzes Signal dazu führt, dass sich Zellen teilen. Dagegen führt ein langes Signal zum Tod der Zelle, oder zu Differenzierung in einen anderen Zelltyp. Wenn der Signalweg angeschaltet wird, führt das (über Zwischenstationen) zu Transkription bestimmter Gene, die dann entsprechend die Zelle verändern. Wie kann die Zelle nun zwischen „kurzen“ (1-2 Stunden) und „langen“ (länger als 4 Stunden) Signalimpulsen unterscheiden? Wie oben beschrieben: ein Mensch würde einfach eine Stoppuhr nehmen, eine Zelle muss das mit den biophysikalischen Eigenschaften ihrer Moleküle erledigen.
In unserer Publikation zeigen wir, dass die Stabilität einer Klasse von RNA-Molekülen den Unterschied zwischen „kurz“ und „lang“ feststellen kann. Viele Gene, die nach Aktivieren des Signalweges transkribiert werden, produzieren instabile RNAs. In einer Computersimulation zeigen wir, dass sie auf kurze und lange Impulse ähnlich reagieren. Wenn aber die produzierte RNA sehr stabil ist, dann akkumuliert sie bei langen Impulsen. Bei kurzen Impulsen hingegen nützt das nichts, weil die RNAs nur für eine kurze Zeit produziert werden, können es bei weitem nicht so viele werden. In der Simulation haben wir gesehen, dass durch diese relativ höhere Akkumulation stabile RNAs zwischen einem kurzen und einem langen Signalimpuls unterscheiden können.
Experimentell konnten wir diese Theorie dann bestätigen, indem wir die Stabilität der RNA-Moleküle von ungefähr zehntausend Genen gemessen haben (insgesamt gibt es in menschlichen Zellen ungefähr zwanzigtausend bis dreißigtausend Gene, viele davon sind aber nicht oder nur sehr wenig aktiv). Mit diesen Daten haben wir die mit der Computersimulation gefundene These bestätigt, nämlich dass es eine Klasse von Genen gibt („immediate late genes“, ILG), die wie „immediate early genes“ sofort inaktiviert werden, deren RNAs aber auf Grund der hohen Stabilität über die Zeit akkumulieren und damit den Unterschied zwischen einem kurzen und einem langen Impuls decodieren können. Viele dieser ILG-Gene befördern den Zelltod, was den oben beschriebenen Effekt erklärt, dass lange Signalimpulse eben den Zelltod herbeiführen.
Über unseren neusten Artikel: Detecting actively translated open reading frames in ribosome profiling data gibt es einen so guten Artikel von Josef Zens, Kommunikationschef des MDC, dass ich gar nichts mehr schreiben muss: https://insights.mdc-berlin.de/de/2015/12/dem-ribosom-bei-der-arbeit-zuschauen/
Wenn ich gefragt werde, was wir den ganzen Tag so treiben im Labor, antworte ich manchmal „wir forschen an post-transkriptioneller Genregulation“. Das klingt so schneidig, dass dem Gegenüber erstmal die Luft wegbleibt, und er oder sie vor Ehrfurcht und Bewunderung erstarrt. Ziel erreicht, und ich nutze den Moment, um mich aus dem Staub zu machen, bevor ich mich erklären muss.
Neugierig geworden? Gut! Ausgangspunkt dieses Beitrages ist das „Zentrale Dogma der Molekularbiologie“, das das Verhältnis zwischen drei wichtigen biologischen Molekülarten beschreibt: die DNA (Erbgut, vor allem dauerhafter Informationsspeicher), die RNA (u.a. temporärer Informationsspeicher) und die an Hand der Information aus der RNA produzierten Proteine (Eiweiße, übernehmen biologische Funktionen wie bspw. Umwandlung von Nahrung in Energie für den Körper). Ursprünglich ging es beim „Dogma“ um Informationsfluss, wie hier (Abschnitte 4 bis 7) gut beschrieben, hat es sich aber zu einer Begründung für einen reduktionistischen Ansatz in der molekularbiologischen Forschung gewandelt. Kurz zusammengefasst: Information, bspw. was eine Zelle zu tun hat in einer bestimmten Situation, fließt nur „hinauf“, d.h. von DNA in Richtung RNA und Protein, und weiter zu Zelle und Gewebe. Wie sich aber in den letzten Jahren mehr und mehr gezeigt hat, und darum geht es grundsätzlich ebenfalls in unserer Publikation, sind solche linearen Vorstellungen nicht korrekt. Vielmehr bewegt sich biologische Information in extrem komplexen Netzwerken, von denen jetzt noch nicht gesagt werden kann, inwieweit sie überhaupt erfassbar sind.
Nun aber zu den konkreten Resultaten in der neusten Studie meines Labors am MDC Berlin, „RC3H1 post-transcriptionally regulates A20 mRNA and modulates the activity of the IKK/NF-κB pathway“. Es geht um das Protein Roquin (auch RC3H1 genannt), das einige Tausend verschiedene RNA-Moleküle binden kann. Zur Orientierung: in menschlichen Zellen gibt es gut 20’000 verschiedene Gene und eine ähnliche Anzahl verschiedener RNAs. Von den einzelnen RNA-Molekülen gibt es zwischen eines (oder gar keines) und einigen Tausend pro Zelle, je nachdem wieviel davon gebraucht wird. RNA-Moleküle, die die Information etwa für ständig gebrauchte Stoffwechsel-Proteine enthalten, sind viel häufiger als solche, die nur kurzzeitig für bestimmte Situationen gebraucht werden. Roquin bindet RNAs der zweiten Art, spezifisch solche, die als Antwort auf DNA-Schäden und auf TNFα gebildet werden. Während DNA-Schäden bspw. durch UV-Strahlen entstehen, und oft den Anfang von Krebserkrankungen bilden, ist TNFα insbesondere an Entzündungen beteiligt. Die Experimente haben gezeigt, dass Roquin von den vier möglichen „Buchstaben“ der RNA (A, C, G, U/T) insbesondere an U bindet, bspw. an die Sequenzen UUUAUUU oder UUUUUAA. Die Bindung von Roquin an RNA führt zu derem schnellen Abbau.
Warum ist das wichtig? Wenn die DNA einer Zelle beschädigt ist oder eine Entzündung stattfindet, führt dies zu einem „Alarmzustand“: die Zellteilung wird gestoppt, Reparaturmechanismen in Gang gesetzt. Konkret bedeutet dies u.a. die Produktion neuer RNAs und der daraus entstehenden Proteine. Einerseits muss nun das Ausmaß dieses „Alarmzustandes“ reguliert werden, andererseits die Zelle anschließend wieder in den „Normalzustand“ zurückgeführt werden. Roquin übernimmt nun genau diese Aufgabe, in dem es solche „Alarm-RNAs“ abbaut.
Eine der RNAs, die von Roquin gebunden werden, codiert das Protein A20. Dieses Protein hemmt den NF-kB-Signalweg, der u.a. von TNFα aktiviert wird. Indem Roquin die RNA von A20 abbaut, gibt es weniger A20-Protein, und damit ist der NF-kB-Signalweg stärker aktiv. Wichtig ist das, wie kürzlich gezeigt wurde, bei rheumatoider Arthritis: Mäuse ohne A20 entwickelten eine ähnliche Form von Arthritis. Wenn A20 also stabilisiert wird, könnte das in gewissen Fällen durchaus eine positive Auswirkung auf Entzündungskrankheiten haben. Unsere Arbeit gibt einen ersten Hinweis wie das gehen könnte: Wenn die Bindungsstelle von Roquin an die A20-RNA blockiert wird, gibt es etwas mehr A20, und das könnte zur Linderung der Arthritis beitragen. Mit dieser Arbeit haben wir einen kleinen Ausschnitt aus den komplexen biologische Netzwerke untersucht, und vielleicht auch einen Hinweis gefunden, wie bestimmte Krankheiten entstehen und möglicherweise sogar behandelt werden können.
